Production of a a phage lambda library of genomic DNA
Before everything begins: prepare solutions and buy kits...
The kits:
- DIG High Prime DNA Labeling and detection Starter Kit II, Roche, cat# 11 585 614 910
- Lambda FIX II/Xho I Partial Fill-In Vector Kit / Gigapack® III XL packaging Extract Stratagène, cat# 248712.
- Klenow Fill-In Kit, Stratagene, cat# 200410
- Wizard® SV Gel and PCR Clean-Up System, Promega, cat# A9281
Biological reagents and Solutions:
- TE buffer, pH 8 (1 mM EDTA pH 8.0, 10 mM Tris-HCl pH 7,5): 50 ml
- 10X STE buffer (1 M NaCl, 200 mM Tris-HCl, pH 7.5, 100 mM EDTA, pH 8.0): 50ml
- SM buffer (290 mg NaCl, 100mg MgSO4-7H2O, 2.5 ml Tris-HCl (1M, pH 7.5), 250 µl gelatin 2 %): 50 ml
- Luria-Bertani (LB) agar: 1L for 12 Petri dishes Ø132 mm (25 g LB Broth pH 7.5 completed with 1L milliQ water and adjust to a pH 7 with 1M HCl). Autoclave.
- LB top agar: 250 ml per 50 reactions (6.25g LB Broth pH 7.5 completed with 250 ml milliQ water and adjusted to a pH 7 with 1M HCl +1.75g agarose for DNA gels). Autoclave
- DNAzol reagent 1 ml (Invitrogen, cat# 10503-027)
- 100% ice-cold ethanol 500 µl (Stored at -20°C)
- 75% ethanol 2 ml
- Phenol-chloroform-isoamide 200 ml (Invitrogen, cat# 15593-031) (Stored at 4°C)
- Chloroform 400 ml (Prolabo cat# 22711-290)
- Agarose
- Tris Borate EDTA (TBE) (0.045M Tris Borate, 0.01M EDTA)
- Sau3AI enzyme (10U/µl) with buffer and BSA (cat# R6191, Promega). (Stored at -20°C)
- Fill-in buffer 10X (60 mM Tris-HCl, 60 mM NaCl, 60mM MgCl2, 0,5% gelatine, 10 mM dithiothreitol, Stratagene). (Stored at -20°C), Provided with the Klenow Fill-In Kit
- Klenow polymerase (5U/µl, Stratagene). (Stored at -20°C), Provided with the Klenow Fill-In Kit
- dATP (10mM, pH 7.5, Stratagene). (Stored at -20°C), Provided with the Klenow Fill-In Kit
- dGTP (10mM, pH 7.5, Stratagene). (Stored at -20°C), Provided with the Klenow Fill-In Kit
- T4 DNA ligase (500U/µl) with buffer (Promega cat# M1804). (store at -20°C)
- pUC19/BamHI –digested control DNA (1µg/µl) provided with the Stratagene Lambda Fix II/XhoI Partial Fill-in Vector kit (Stratagene). (Stored at -20°C)
- pMF DNA (0,5µg/µl) (Stored at -20°C), provided with the Stratagene Lambda Fix II/XhoI Partial Fill-in Vector kit (Stratagene).
- XL1-MRA (P2), Gigapack III XL provided with the Stratagene Lambda Fix II/XhoI Partial Fill-in Vector kit (Stratagene), stored in glycerol (4.1%) at -80°C.
- LB medium supplemented with 10mM of MgSO4 and 0.2% of L-maltose. Autoclave
- 1M MgSO4: 50ml
- 20% of L-maltose: 50ml
- DMSO
- dNTP mix (Promega, cat# U1511) (stored at -20°C)
- GoTaq® polymerase and buffer (Promega, cat# M7650) (stored at -20°C)
- EcoRI enzyme with buffer H and BSA (Promega, cat# R6011) (stored at -20°C)
- Nylon membrane for colony and plaque hybridisation (Roche, cat# 1169908300)
- Wizard Plus SV Minipreps DNA Purification System (Promega, cat# A9281)
- Denaturation solution (0.5M NaOH, 1.5M NaCl) : 1L (stored at Room temperature)
- Neutralisation solution (1.5M NaCl, 1 M Tris-HCl, pH 7.4) : 1L (stored at Room temperature)
- 2X SSC (0.3M NaCl, 30mM sodium citrate, pH 7: 1L (stored at Room temperature)
- 2X SSC and 0.1% SDS:1L (stored at Room temperature)
- 0.5X SSC and 0.1% SDS:1L (stored at Room temperature)
- 0.2X SSC and 0.1% SDS:1L (stored at Room temperature)
- Washing buffer (0.1 M maleic acid + 0.15 M NaCl, adjust with solid NaOH to pH 7.5 at 20°C, 0.3 % Tween 20): 1L (freshly prepared)
- Maleic Acid Buffer: 0.1M Maleic Acid, 0.15M NaCl, adjust with NaOH (solid) to pH7.5 (20°C) (approximately 15.8g of NaOH pills) (stored at Room temperature
- Blocking solution: provided with the DIG High Prime DNA Labeling kit (freshly prepared)
- Antibody solution provided with the DIG Hich Prime DNA Labeling kit (freshly prepared)
- Detection buffer (0.1M Tric-Hcl, 0.1M NaCl, pH9.5 (20°C) (stored at Room temperature)
- Amersham hyperfilm ECL (GE Healthcare, cat# 2890 6840).
- Water
- Dounce
- Whatman paper
Genomic DNA isolation
For each enzymatic digestion, genomic DNA (gDNA) was extracted from 300 adult flukes of Schistosoma mansoni. The extraction was done with the DNAzol kit (Invitrogen). For each reaction, 1 ml of DNAzol was added to adult flukes in a 1.5 ml Eppendorf tube. Flukes were suspended by pipetting the reaction up and down. This mix was transferred into a Dounce (on ne connait pas le type) and tissues were lyzed by 10 up and down movements.
The lysate was centrifuged for 4 min at 10 000g at 4°C. The supernatant was transferred into a fresh 2 ml tube and was again centrifuged to eliminate the remaining cell debris. The supernatant was transferred into a new 1.5 ml tube.
DNA was precipitated by addition of 500 µl 100% ice-cold ethanol and the mix was homogenized by inversion for 1 to 3 mins, centrifuged for 2 mins at 4000g at 4°C and the supernatant was discarded. The pellet was washed with 1 ml 75% ethanol and centrifuged at 7000 rpm for 2 mins at room temperature. The pellet was eluted into 200µl milliQ water on ice bath for 10 mins.
A phenol-chloroform step was finally performed to purify the DNA. An equal volume of phenol-chloroform was added and the content of the tube was mixed by gentle inversion until the pellet was dissolved. The tube was centrifuged at 10 000 rpm for 10 mins at room temperature. The upper aqueous layer was transferred to a new tube. To eliminate any residual phenol, an equal volume of chloroform was added to this phase and the mix was centrifuged at 10000 rpm for 5 mins at room temperature. The supernatant was collected for the rest of the protocol. The phenol phase was kept as we observed some remaining DNA at the white layer that remains on the surface. This remaining DNA was eluted from the phenol layer by addition of 200 µl milliQ water and phenol-chloroform extracted as explained above.
The quantity of DNA was evaluated by spectrophotometer (approximately 2.5 mg in a final volume of 400 µl). 5 µl was loaded on a 0.7 % agarose gel to evaluate integrity by visual inspection as illustrated in figure 1.
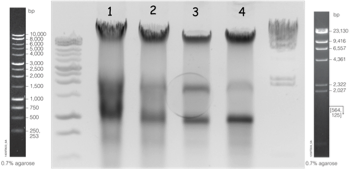
Fig. 1: Extraction of gDNA from Schistosoma mansoni strain. Lanes 1 and 3: first and second extraction of the GH2 S. mansoni strain. Lanes 2 and 4: first and second extraction of the BRE S. mansoni strain.
Digestion and fill-in
Partial digestion:
The next steps consisted in generating compatible overhangs to the cloning vector: the Stratagene Lambda Fix II vector which is predigested with XhoI and filled in with two nucleotides, leaving 3’-CT-5’ overhangs. Therefore, we performed a partial digestion with Sau3AI (10 U/µl, Promega) and a fill-in with two nucleotides to generate 5’-GA-3’. This digestion step had to be optimized to generate fragments spanning 10 to 23 kb. The result of the partial digestion relied on the quantity and quality of gDNA. Therefore, this optimization of the digestion conditions had to be optimized each time a new gDNA extract was prepared. This optimization was performed in two steps.
A first digestion was performed using increasing concentration of enzymes: 1x10-1, 1x10-2, 1x10-3, 1x10-4 Units of enzymes per μg of gDNA. The appropriate range that generated bands between 10 to 23 kb was chosen for further narrow digestion. The reaction was performed during 15 min at 37°C and inactivated for 15 mins at 65°C. An example of such a digestion on a gDNA preparation at 6.5 μg/μl is illustrated on figure 2 and reaction mixes were prepared as outlined in table 1.
Table 1: Partial digestion at restrict scale of enzyme concentration. The quantity are given in μl
*1: The enzyme is prepared doing serial dilution in the dilution mix containing 10X buffer B, 100X BSA and fill up with milliQ water.
1
|
2
|
3
|
4
|
5
|
C+
|
|
No digestion
|
1.10-4U/μg of gDNA
|
1.10-3U/μg of gDNA
|
1.10-2U/μg of gDNA
|
1.10-1U/μg of gDNA
|
1.10-1U/μg of gDNA
|
|
gDNA (28µg)
|
5
|
5
|
5
|
5
|
5
|
5
|
Buffer B 10X
|
3
|
3
|
3
|
3
|
3
|
3
|
BSA 100X
|
0.3
|
0.3
|
0.3
|
0.3
|
0.3
|
0.3
|
Water
|
21.7
|
18.5
|
18.5
|
18.5
|
18.5
|
18.5
|
Sau3AI (10U/µl) *1
|
0
|
3.25 (105X dilution)
|
3.25 (104X dilution)
|
3.25 (103X dilution)
|
3.25 (102X dilution)
|
3.25 (10X dilution)
|
Final volume
|
30
|
30
|
30
|
30
|
30
|
30
|
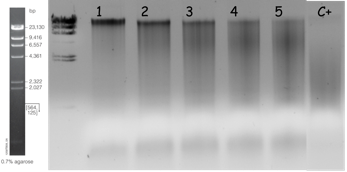
Fig. 2: Partial digestion of gDNA with increasing concentration of Sau3AI. The reaction conditions are explained in Table 1. Digestion in the positive control (C+) was performed during 1 hour. Migration was performed using 25µl of digested samples on 0.7% agarose gel in TBE at 100V for 1h30 at room temperature.
A thorough digestion was then performed in the appropriate range using increasing concentration of enzymes increased twice in between each reaction condition. Each reaction condition was performed 10 times, each time in 30 µl. The 10 fractions were pooled and an aliquot was loaded and size separated on a 0.4% gel agarose by electrophoresis. This allows to chose the condition that was optimal for generation of the 10 to 23 kb bands. An example of such a digestion on a gDNA preparation at 6.5 μg/μl is illustrated on figure 3 and reaction mixes were prepared as outlined in table 2. In this example, sample 2 was chosen for further step.
Table 2: Partial digestion at restrict scale of enzyme concentration.
1
|
2
|
3
|
4
|
5
|
C+
|
|
Concentration (U/µg of gDNA)
|
No digestion
|
1.10-4
|
2.10-4
|
4.10-4
|
6.10-4
|
6.10-4
|
gDNA (65,5µg)
|
5
|
50
|
50
|
50
|
50
|
5
|
Buffer 10X
(NEB B)
|
3
|
30
|
30
|
30
|
30
|
3
|
BSA 100X
|
0,3
|
3
|
3
|
3
|
3
|
0,3
|
Water (µl)
|
21.7
|
214
|
213
|
212
|
211
|
21.5
|
Sau 3AI (10U/µl)
|
0
|
2.62
|
3.92
|
5.24
|
6.55
|
0.65
|
Final volume
|
30
|
300
|
300
|
300
|
300
|
30
|
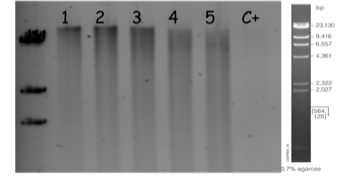
Fig. 3: Partial digestion of gDNA with increasing concentrations of Sau3AI. The reaction conditions are explained in Table 2. Positive control (C+) was digested for 1 hour. Migration was performed using 15 µl of digested samples on 0.4% agarose gel in TBE at 18V for 12h at 4°C.
Partial fill-in
Digestion reactions that produced fragments of expected size were purified doing a phenol-chloroform as previously detailed. The upper layer (approximately 300 µl) was collected into a fresh tube and used for the next steps of the protocol.
We estimated that the samples contain approximatively 50 µg of gDNA in a volume of 300 µl. The templates were partially filled-in as described in the table 3. The reactions were incubated in a dry bath at 22°C for 15 mins. To confirm that enzymatic activity was present in the solution, a plasmid (pMF) was used as a positive control. The reaction was stopped by addition of 150 µl 1X STE buffer and 50µl 10X STE buffer (See the preparation of reagents). The control condition was stopped only by addition of 475 µl 1X STE buffer. The final volumes of all samples were 500 µl.
Table 3:
*1: Fill-in buffer 10X containing 60 mM Tris-HCl, 60 mM NaCl, 60mM MgCl2, 0.5% gelatine, 10 mM dithiothreitol.(Stratagene)
*2:pMF DNA (0.5µg/µl) included in the Gigapack III XL
Sample ≈ 50µg
|
Control pMF*2 (0.5 µg/µl)
|
|
DNA
|
257
|
3
|
Fill-in buffer*1 10X
|
30
|
2.5
|
dATP (10mM, pH 7.5)
|
5
|
1
|
dGTP (10mM, pH 7,5)
|
5
|
1
|
Water
|
0
|
16.5
|
Klenow polymerase (5U/µl, Stratagene)
|
3
|
1
|
Final volume
|
300
|
25
|
Samples were then purified using an equal volume of phenol-chloroform. All reactions were centrifuged for 2 mins at 10 000 rpm at room temperature and the upper aqueous layer was transferred into a fresh tube. The phenol traces were eliminated with an equal volume of chloroform. The tubes were gently inverted several times and centrifuged. The upper layer was collected into a fresh tube.
DNA was precipitated by addition of 2 volumes of 100% ethanol (1ml) and incubated at -20°C for 30 min. Samples were then spinned down at 4°C for 10 mins at 13 000 rpm. DNA pellets were then washed with ice-cold 70% ethanol (1 ml) and air-dried. DNA was suspended in 3.5 µl TE buffer for the control reaction and in 10 µl TE buffer for the sample reactions. 1µl of samples were then size separated on a 0.7% agarose gel in order to control the size and the quantity of the partially digested gDNA (figure 4).
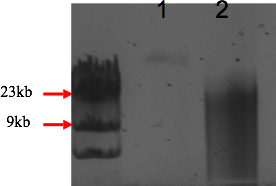
Fig. 4: Partial fill-in of gDNA with the Klenow Fill-In kit (Stratagene, La Jolla, CA). The reaction was performed as outlined in Table 3. Migration was performed using 1µl of filled-in sample and 1 µg of pUC19 DNA on a 0.7% agarose gel in TBE at 100V for 2h at room temperature. (1) plasmid control, (2) filled in sample.
Ligation into the lambda vector
The ligation of the insert into the vector was performed as recommended in the lambda FIX II/Xho I partial Fill-In Vector Kit (Stratagene) using 0.4 μg of insert for every 1 μg of vector. The quantity of gDNA insert was estimated according to the visual inspection of the gel agarose electrophoresis previously performed (see figure 4). The ligation was performed at 4°C for 15 h as described in the table 5.
It was possible to control the ligation procedure using a pUC19/BamHI –digested control DNA provided with the kit and the previously prepared fill-in control pMF. It was expected that the pUC19 would ligate to himself and generate 3 bands on the gel on the contrary to the fill-in control pMF was expected to migrate as a single band on the gel. Control reactions were outlined in Table 6.
Table 6: Reaction mix for ligation and self-ligation (control)
*1:T4 ligase buffer (Promega).
*2:T4 DNA ligase (400U/µl, Promega).
*3: Lambda FIX II vector/Xho I partially filled-in, (Stratagene)
Sample
|
Cloning control pMF
|
|
DNA
|
0.4 μg
|
0.8
|
T4 buffer*1
|
0.5
|
0.5
|
T4 ligase*2
|
0.5
|
0.5
|
Water
|
2.5
|
2.2
|
λ FIX vector*3
|
1 μg
|
1
|
Final volume
|
5
|
5
|
Packaging
The recombinant lambda phage (gDNA insert + phage vector) were packaged into the phage capsid using the commercially available Gigapack III XL packaging extract (Stratagene), which preferentially package very large insert (47-51 kb recombinant). 1 to 4 µl (containing 0.1 to 1.0 µg of ligated DNA) were added to a vial of the Stratagene kit and were placed into a dry bath for 2 h at 22°C. The reaction was stopped with 500 µl SM buffer and the packaging solution was sterilized by addition of 20 µl chloroform. The content of the tube was briefly centrifuged and the supernatant was transferred into a new tube in sterile condition. This solution with the packaging products can be stored at 4°C for 1 month.
Bacterial cultures for transformation by the recombinant phage, titration and amplification of the library.
Bacterial culture:
A liquide culture of the freshly streaked lysogenic E.coli (XL1-MRA P2, Gigapack III XL) was grown to exponential phage into LB medium supplemented with 10mM of MgSO4 and 0.2% of L-maltose at 37°C. The culture was centrifuged for 10 min at 500 g and suspended in 10mM MgSO4 to an OD of 0.5. This suspension was used for transformation by the encapsulated phage, titration and library amplification.
Titration of the library:
The efficacy of encapsulation of recombinant phage containing the gDNA library was evaluated by transforming an E.coli strain provided by the manufacturer. 1µl of the packaged extract was added to 200 µl of the freshly prepared E.coli (XL1-MRA P2) (see previous section) and was incubated at 37°C for 15min.
4.5 ml of pre-warmed (48°C) LB Top agar was added to the transformed bacterial strain and immediately poured on the pre-warmed (37°C) LB agar plates. The plates were cooled at room temperature for several minutes and incubated upside down at 37°C for 15 h. Phage forming units were counted after overnight incubation. Plates can be stored at 4°C for several weeks.
Genome coverage estimation:
(Total number of pfu * mean size of insert (15kb)) / (genome size)
Amplification of the library:
5.104 pfu of recombinant packaged phages were incubated with 450µl of freshly prepared E.coli (XL1-MRA P2) and incubated at 37°C for 15 mins. 4.5 ml of pre-warmed (48°C) LB Top agar was added to the transformed bacterial strain and immediately poured on the pre-warmed (37°C) LB agar plates. The plates were cooled at room temperature for several minutes and incubated upside down at 37°C for 8h. Then, each Petri dish was suspended in 8ml of SM buffer and left overnight under gentle rocking agitation at 4°C. The SM buffer was collected in a 15ml falcon tube . Each dish was then rinsed once with 2 ml of SM buffer that were added to the previous 8 ml. 5% chloroform (final volume) were added to the 10 ml collected phages. The tubes were incubated for 15 mins at room temperature and centrifuged for 10 mins at 500g. The upper aqueous layer was placed into a fresh tube and samples were stored with 0.3% chloroform (final volume) at 4°C or into 7% DMSO (final volume) at -80°C.
This amplified library was titled as previously described.
Screening
The library was screened for the SmPoMuc genes which is a multifamily genes containing highly repeated units in their 5’ part. We used a probe (UR1) that corresponds to the intronic sequence that spans the region in between two repeat units of the SmPoMuc genes. The probe was labeled with the DIG High Prime DNA Labeling and Detection Starter Kit II using Random primed DNA labeling with digoxigenin-dUTP, alkali-labile and chemiluminescence with CSPD (Roche, cat# 11 585 614 910).
Synthesis of the probes:
The probe (UR1) was generated by PCR. PCR conditions are outlined in Table 7. The probe was gel purified using the Wizard® SV Gel and PCR Clean-Up System (Promega) quantified and labeled with the DIG High Prime DNA Labeling kit (Roche) according to the manufacturer instruction: 1µg (contained in 16µl of milliQ water) was denatured by heating at 99°C for 10 minutes and immediately placed on ice. 4µl of DIG High Prime reagent (vial 1) was added and this mix was incubated for 24h at 37°C. The reaction was stopped by addition of 2µl EDTA (0.2M, pH=8).
Table7: PCR conditions for probe amplification
1 mix
|
PCR program
|
|
gDNA (10ng/µl)
|
1 µl
|
Step 1: 1min at 95°C
Step 2: 30s at 95°C
Step 3: 30s at 60°C
Step 4: 30s at 72°C
Step 5: go to step 2, 30 fold
Step 6: 10min at 72°C
Step 7: HOLD at 4°C
|
5X goTaq buffer (Promega)
|
6 µl
|
|
dNTP (10mM)
|
0.6 µl
|
|
forward primer (BF2) (10µM)
|
0.6 µl
|
|
reverse primer (BR2) (10µM)
|
0.6 µl
|
|
Water
|
21.05 µl
|
|
goTaq polymerase (Promega)
|
0.15 µl
|
|
Final volume
|
30 µl
|
|
Primer sequences
|
||
BF2
|
TCTCACATTTCAGGTGACCTC
|
|
BR2
|
AACTCACCTGTGGGTTTGTCTG
|
|
We estimated that 1 µg of PCR product generates 2.3 µg of labeled probe. This estimation was performed doing a serial dilution of the labeled probe and comparing this dilution to a DIGH labeled control DNA as detailed in the instruction manual of the kit..
Performing plaque lift (Transfer to membranes):
2.104 pfu of recombinant packaged phages were incubated with 450µl of freshly prepared E.coli (XL1-MRA P2) and incubated at 37°C for 15 mins. 4.5 ml of pre-warmed (48°C) LB Top agar was added to the transformed bacterial strain and immediately poured on the pre-warmed (37°C) LB agar plates (135 mm). The Petri dishes were incubated overnight at 37°C. The dishes were then cooled for 2h at 4°C before the transfer on the membrane or stored at 4°C until the transfer. The nylon membrane disc (Roche, cat# 1169908300) was left on the agar plates for 3 to 5 min. The exact position of the membrane on the Petri dish was noticed. Using tweezers, the membrane disc was then removed and placed with the DNA side up on Whatman paper for 1 min. Three sheets of Saran wrap were spread on the bench and 4 ml of denaturation solution, neutralization solution and 2X SSC were applied on each wrap. With the DNA facing up, the membrane disc was incubated in denaturation solution for 5 min, in neutralization solution for 8 min and in 2X SSC for 10min. In-between each of this incubation steps, the membrane disc is placed with the DNA side up for 1 min on a new Whatman paper. DNA is finally linked to the membrane by exposure to 120mJ in a UV crosslinker (Fisher Bioblock). The membrane disc can be used directly or stored at 4°C with insert of Whatman paper between two membranes (DNA facing up).
Estimation of optimal hybridization temperature:
Topt= (49.82+0.41(%CG)-(600/lenght of probe))-20 à 25°C=33 à 38°C
For UR1:
Tm= (49.82+0.41(38,19)-(600/1050))=64,8°C
Topt= Tm-20 à 25°C
Prehybridization:
The ref for the roller tubes.
The pre-hybridisation step was performed in 30 ml of DIG easy hyb buffer provided with the DIG High Prime DNA Labeling kit (Roche), for 30 min at 42°C in a hybridization oven. A single membrane disc (135 mm diameter) per roller tube is recommended to lower the background.
Hybridization:
4.4 µl of DIG-labeled probe (1 µg/µl) was used for each membrane. The probe was denatured during 5 min at 99°C and was immediately cooled on ice. The denatured probe was added to 20 ml of DIG easy hyb and poured into each roller tube. Incubation was performed overnight (16-19h) with rotation in a hybridization oven at 42°C.
Stringency washes:
After overnight incubation, DIG easy hyb buffer containing the probe was removed from the roller tubes and washes were performed in the same roller tubes in the hybridization oven. First, two low stringency washes were performed with 200 ml washing solution 1 (2X SSC, 0.1% SDS) for 5 min. Then, two high stringency washes were performed in 200 ml of washing solution 2 (0.5X SSC, 0.1% SDS) for 15 mins at 68°C. Finally, two very high stringency washes were performed in 200 ml of solution 3 (0.2X SSC, 0.1% SDS) for 15 mins at 68°C.
Chemiluminescent detection:
The next steps were performed at room temperature in the roller tubes using the hybridization oven. After hybridization and stringency washes, the membrane was rinsed once in 200 ml of washing buffer for 5 min at room temperature. Then, washing buffer was removed and the membrane was incubated for 30 min in 100 ml of blocking solution. The blocking solution was replaced by antibody solution for 30 min. Two washes were then performed in 200 ml of washing buffer during 15 min. The membrane was finally equilibrated during 5 min with 100 ml detection buffer.
The membrane discs (DNA facing up) were then removed from the roller tubes and placed on a clean glass plate and 1.5 ml of CSDP solution (provided with the kit) was applied on each membrane. The membrane + CSPD were then covered with Saran wrap for 5 min. After incubation, the excess of CSPD was removed using a towel paper and pushing this excess outside of the saran wrap. The glass plate was placed into a X-ray cassette for the revelation on Amersham hyperfilm ECL (GE Healthcare, cat# 2890 6840).
Film exposure was performed during 1 to 3 min depending on the signal intensity.
Secondary and tertiary screening:
The detected spots are reported on the initial Petri dish. Using a 200 µl tips, a plug surrounding the positive area was sampled from the Petri dish. This plug contains the UR1 positive phage but also negative phages and for this reason it was necessary to perform secondary and tertiary screening until a pure phage was obtained. This plug was incubated with 1 ml SM buffer overnight in a 1.5 ml Eppendorf tube under gentle shaking. The SM buffer was removed and placed into a fresh tube. 5% chloroform (final volume) was added to the 1 ml collected phages. The tubes were incubated for 15 mins at room temperature and centrifuged for 10 mins at 500g. The upper aqueous layer was placed into a fresh tube and samples were stored with 0.3% chloroform (final volume) at 4°C or into 7% DMSO (final volume) at -80°C as previously described for the whole library. The presence of the UR1 unit repeat was also checked by PCR on 1ul of the phage solution. PCR conditions are those described in table 7. The secondary and tertiary screening was performed as described for the primary screening. The fraction containing the phage solution enriched for UR1 was plated on 135 mm plates at different dilution from 10-1 to 10-3
For secondary screening, the density of phage recommended is:
For tertiary screening the density of phage recommended is:
The Petri dish are then processed for UR1 detection as previously described.